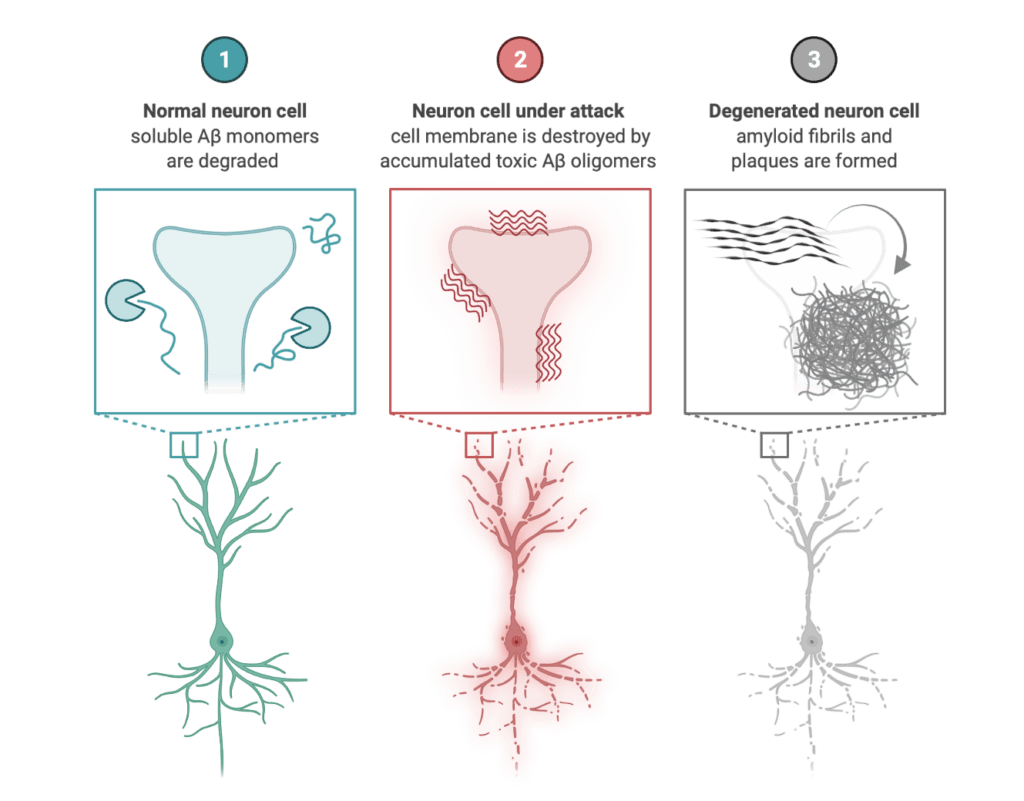
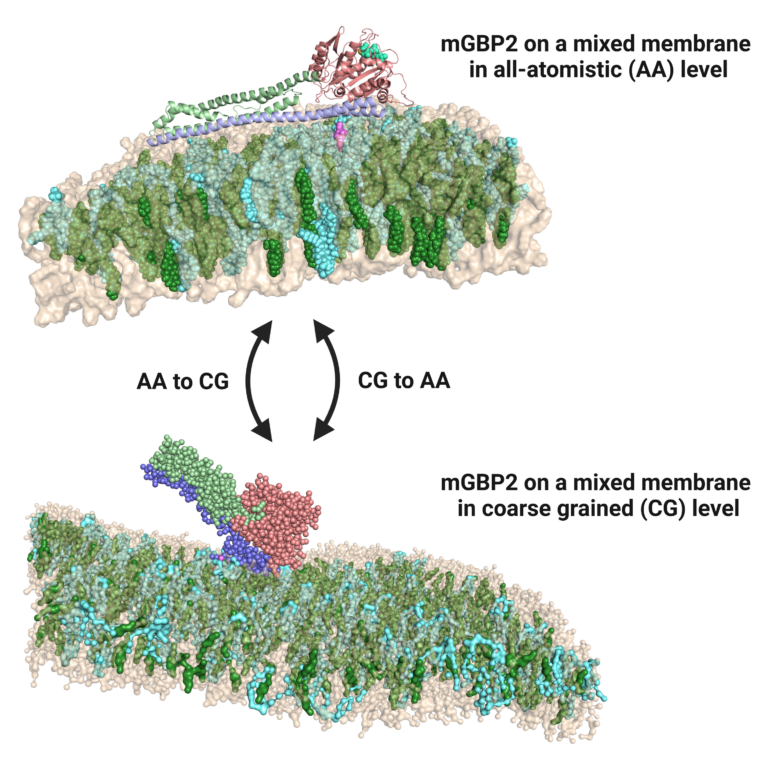
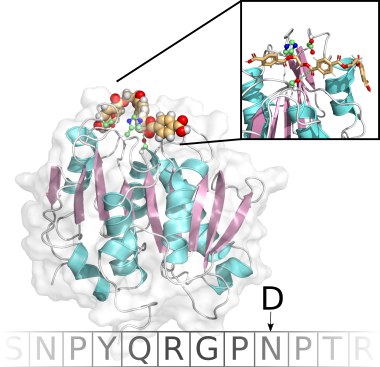
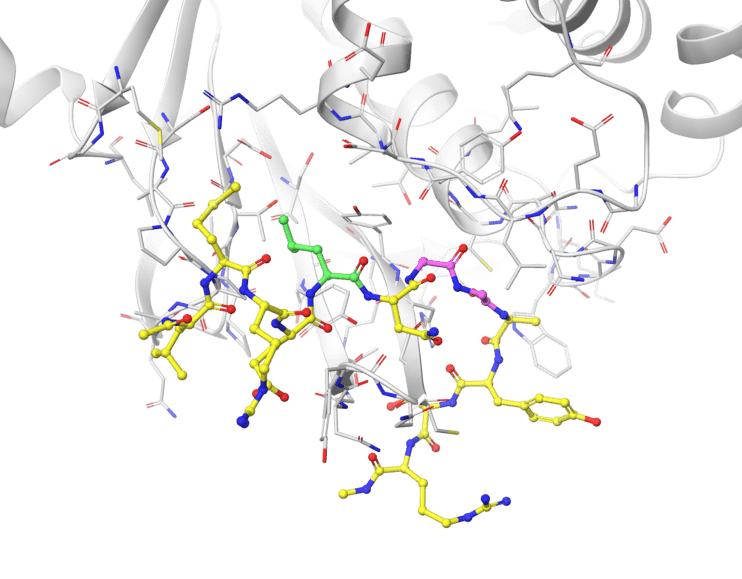
A brief history of amyloid aggregation simulations
H Fatafta, M Khaled, B Kav, OO Olubiyi, B Strodel
Wiley Interdisciplinary Reviews: Computational Molecular Science 14 (1), e1703, 2024
A hairpin motif in the amyloid-β peptide is important for formation of disease-related oligomers
M. Khaled, I. Rönnbäck, LL. Ilag, A. Gräslund, B. Strodel, N. Österlund
Journal of the American Chemical Society 145 (33), 18340-18354, 2023
Transition Networks Unveil Disorder-to-Order Transformations in Aβ Caused by Glycosaminoglycans or Lipids
M. Schäffler, S. Samantray, B. Strodel
International Journal of Molecular Sciences (2023)
Structural dissection of the first events following membrane binding of the islet amyloid polypeptide
L. Khemtemourian, H. Fatafta, B. Davion, S. Lecomte, S. Castano, B. Strodel
Frontiers in molecular biosciences 9, 849979, (2022)
Amyloid-β peptide dimers undergo a random coil to β-sheet transition in the aqueous phase but not at the neuronal membrane
Fatafta H, Khaled M, Sayyed-Ahmad A, Strodel B
Proceedings of the National Academy of Sciences (2021)
Disorder-to-order transition of the Amyloid-beta peptide upon lipid binding
H Fatafta, B Kav, BF Bundschuh, J Loschwitz, B Strodel
Biophysical Chemistry, Volume 280, Pages 106700 (2021)
Different force fields give rise to different amyloid aggregation pathways in molecular dynamics simulations
S Samantray, F Yin, B Kav, B Strodel
Journal of Chemical Information and Modeling, Vol. 60, Pages 6462-6475 (2020)